
I am a Clinical Bioinformatics Lead at Baebies.
At Baebies, I am currently working on implementing scalable clinical NGS data analysis workflows and developing novel computational tools that advance newborn screening and pediatric testing. My research experience spans a wide range of topics, including, gene regulation, computational drug discovery methods, developing analysis pipelines for high-throughput sequencing data, and novel tools for data visualization.
I got my Ph.D. in Human Genetics with a focus in Computational Biology under the supervision of Aleksander Milosavaljevic from the Department of Molecular and Human Genetics at Baylor College of Medicine. Later I did Bioinformatics Postdoctoral Fellowship with Murat Can Cobanoglu at the Lynda Hill Department of Bioinformatics at UT Southwestern Medical Center. I also have a Master's degree in Molecular and Cell Biology from the University of Texas at Dallas and Bachelor's degree in Biochemistry from Rutgers University.
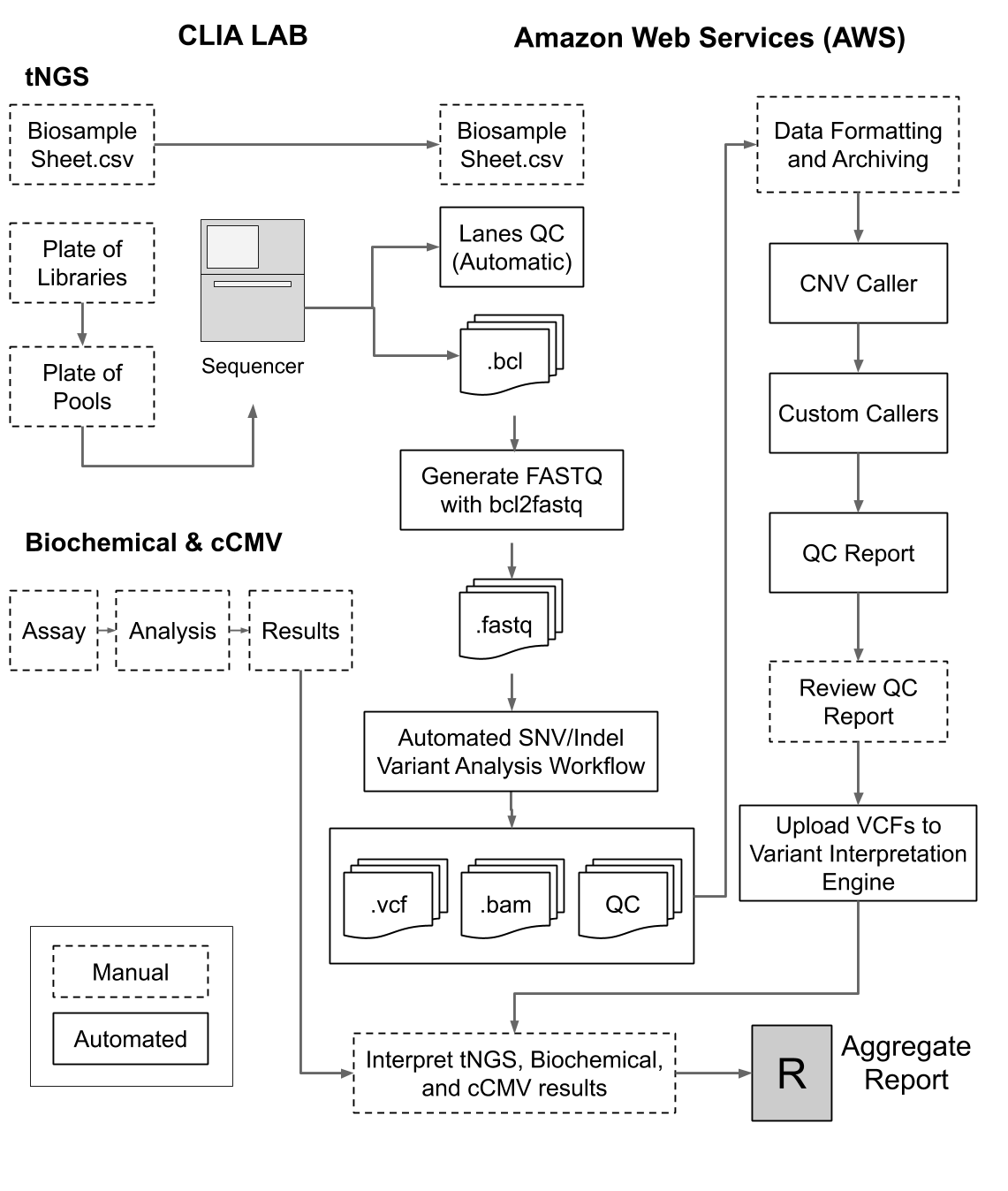
Newborn screening (NBS) initiated by public health laboratories (PHL) in the US and worldwide has significantly reduced childhood morbidity and mortality for inherited conditions. There are several conditions beyond public health screening that may benefit from early identification through NBS but are not yet routinely screened in PHL. To maximize the potential benefit of NBS, we developed an expanded/supplemental NBS service which integrates molecular and biochemical assays, as well as targeted next generation sequencing (tNGS). Panel tests for 32 conditions and risk factors that are not typically covered in all state PHLs. This includes 6 biochemical assays that are followed with 2nd-tier tNGS, 25 additional stand-alone tNGS screens, and 1 qPCR assay. The full test panel may be used either as a physician ordered service, or the tNGS portion of the panel can be used to complement the needs of PHLs.
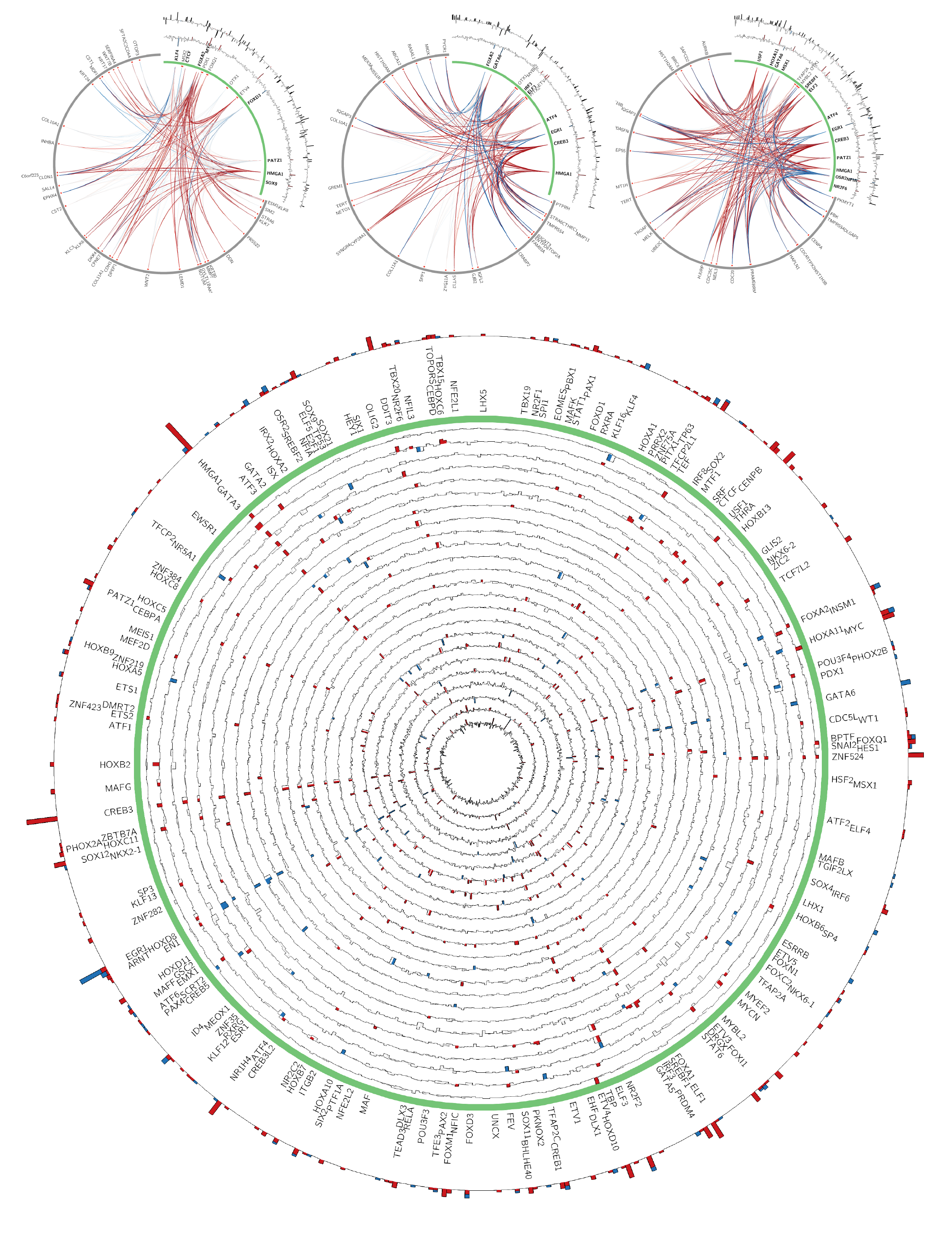
Cancer is a complex disease, and modern sequencing methods offer the potential to unravel that complexity. We work on the problem of understanding the regulators and their downstream targets involved in maintaining or driving the cancer pathology. To uncover the transcriptional dysregulation in cancer, we developed a computational model that takes transcriptomic data and learns differentially regulated genes, as well as potential regulators, based on cell/tissue-specific context. Specifically, we integrate context specific regulatory networks generated across 1000 of cell/tissues types to precondition the model and infer a regulatory network for a given condition using transcriptomic data. We applied our method on the cancer types profiled in TCGA. We show that our method recovers several well-known regulators and identifies potential novel regulators involved in cancer progression.
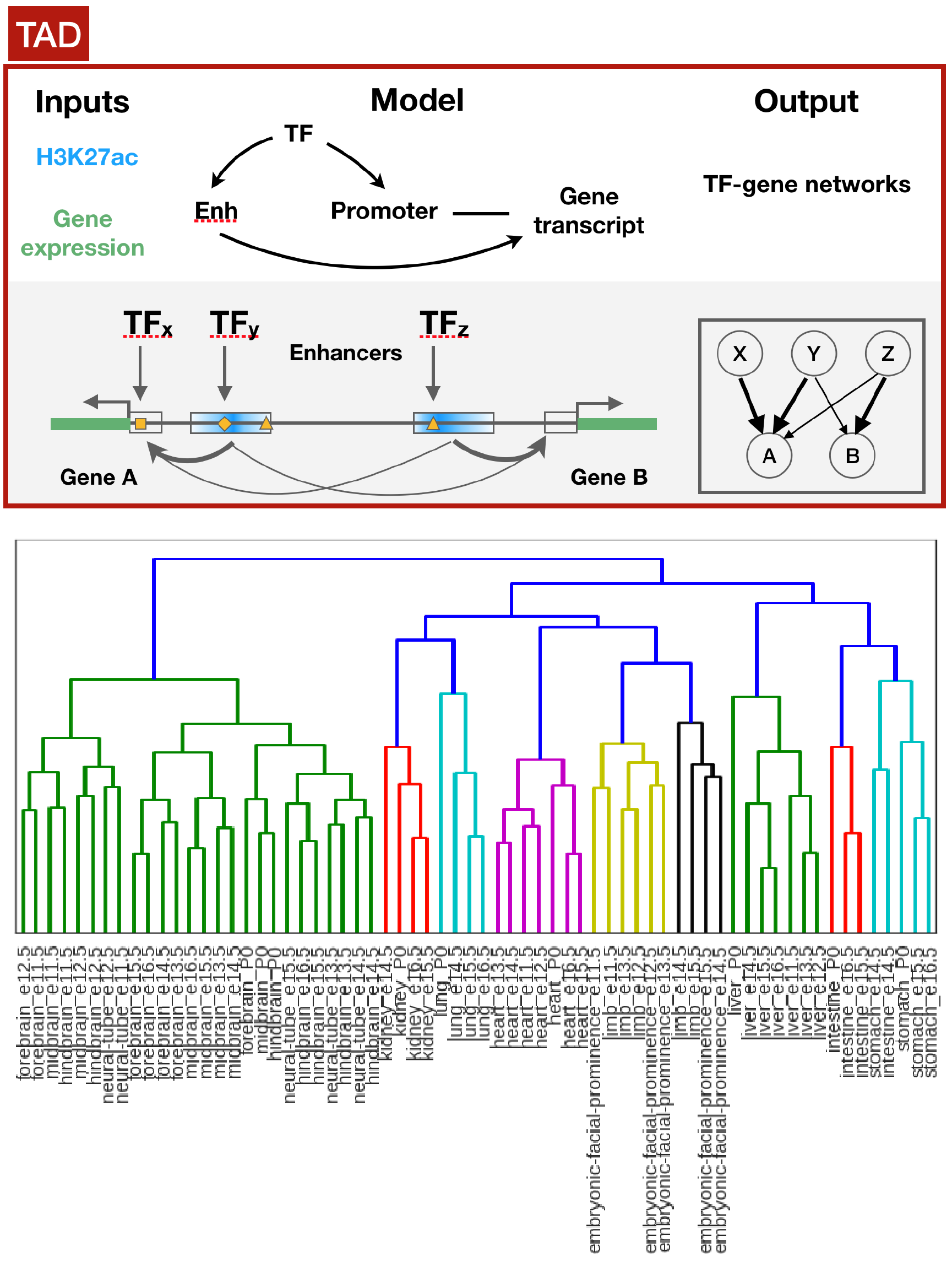
Different groups have started acquiring both transcriptomic and epigenomic datasets to understand gene regulation during cellular differentiation or disease progression. However, integration of epigenomic and transcriptomic datasets are typically done in an ad-hoc manner. Also, there exists no tools or resources that uniformly processed existing epigenomic and transcriptomic datasets. Therefore, we developed a method that integrates epigenomic and transcriptomic data to construct cell/tissue-specific TF-gene regulatory networks. The method takes into account of the topologically associated domains (TAD), evolutionary conservation, and well-curated transcription factor recognition motifs to construct the networks. We applied our method on mouse ENCODE dataset and showed that our method can group cell/tissue-specific regulatory networks based on their cell type of origin and determine novel regulatory interactions that are biologically relevant.
Integrate epigenomic and transcriptomic datasets to construct TAD aware regulatory networks.
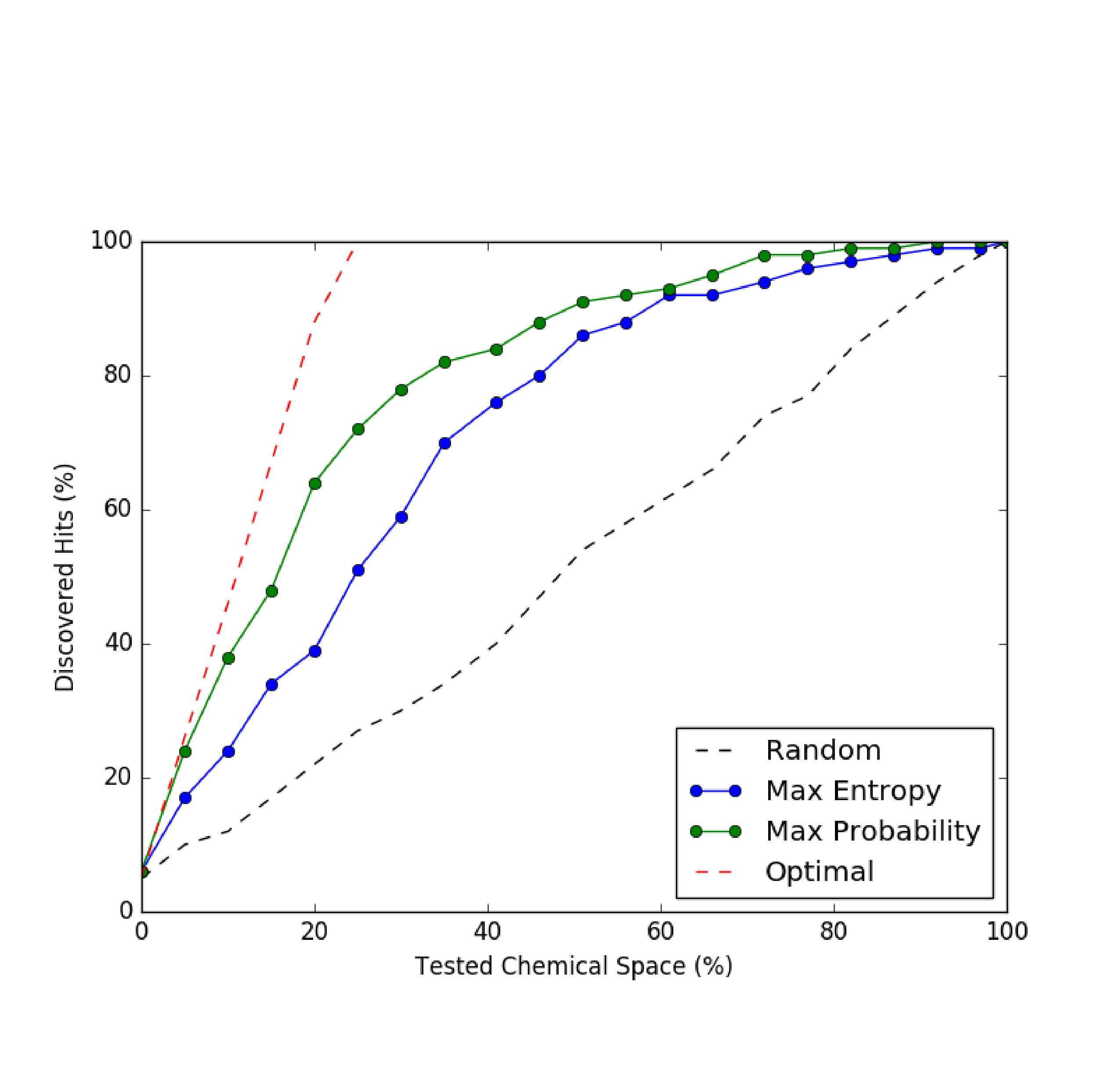
The current approach of finding active compounds is through screening large chemical library on a given target. However, the brute force approach takes time, resources, and do not learn from prior experiments. To overcome the problem, we can use machine learning to learn chemical features from previous experiments that responded to the target. Then we can either explore or exploit the chemical space to find a new set of compounds to test experimentally. Such approach of actively selecting compounds based on results of prior experiments can drastically reduce time and resources to search an active compound. I implemented simple active learning routine that picks compounds with a high chance of responding to the target (code).
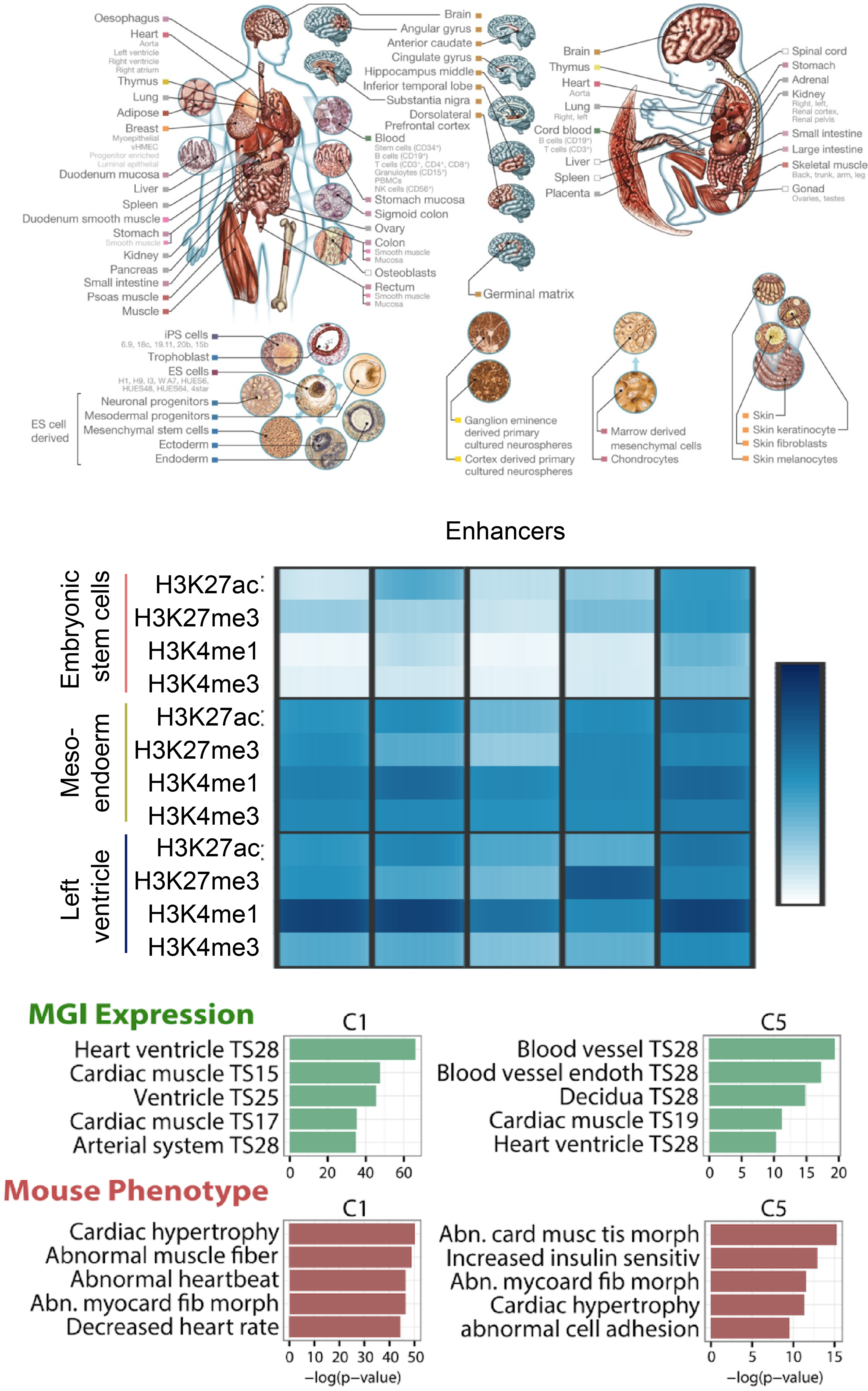
The reference human genome sequence set the stage for studies of genetic variation and its association with human disease, but epigenomic studies lack a similar reference. To address this need, the NIH Roadmap Epigenomics Consortium generated the largest collection so far of human epigenomes for primary cells and tissues. Here we describe the integrative analysis of 111 reference human epigenomes generated as part of the programme, profiled for histone modification patterns, DNA accessibility, DNA methylation and RNA expression. We establish global maps of regulatory elements, define regulatory modules of coordinated activity, and their likely activators and repressors. We show that disease- and trait-associated genetic variants are enriched in tissue-specific epigenomic marks, revealing biologically relevant cell types for diverse human traits, and providing a resource for interpreting the molecular basis of human disease. Our results demonstrate the central role of epigenomic information for understanding gene regulation, cellular differentiation and human disease.
Integrative analysis of 111 reference human epigenomes.
Nature, 518:317-330.
Integrative analysis leads (equal contributors)
A. Yen, A.H. Moussavi, P. Kheradpour, Z. Zhang, J. Wang, M.J. Ziller, V. Amin , J.W. Whitaker, M.D. Schultz, L.D. Ward, A. Sarkar, G. Quon, R.S. Sandstrom, M.L. Eaton, Y.C. Wu, A. Pfenning, X. Wang & M. Claussnitzer
(aticle)
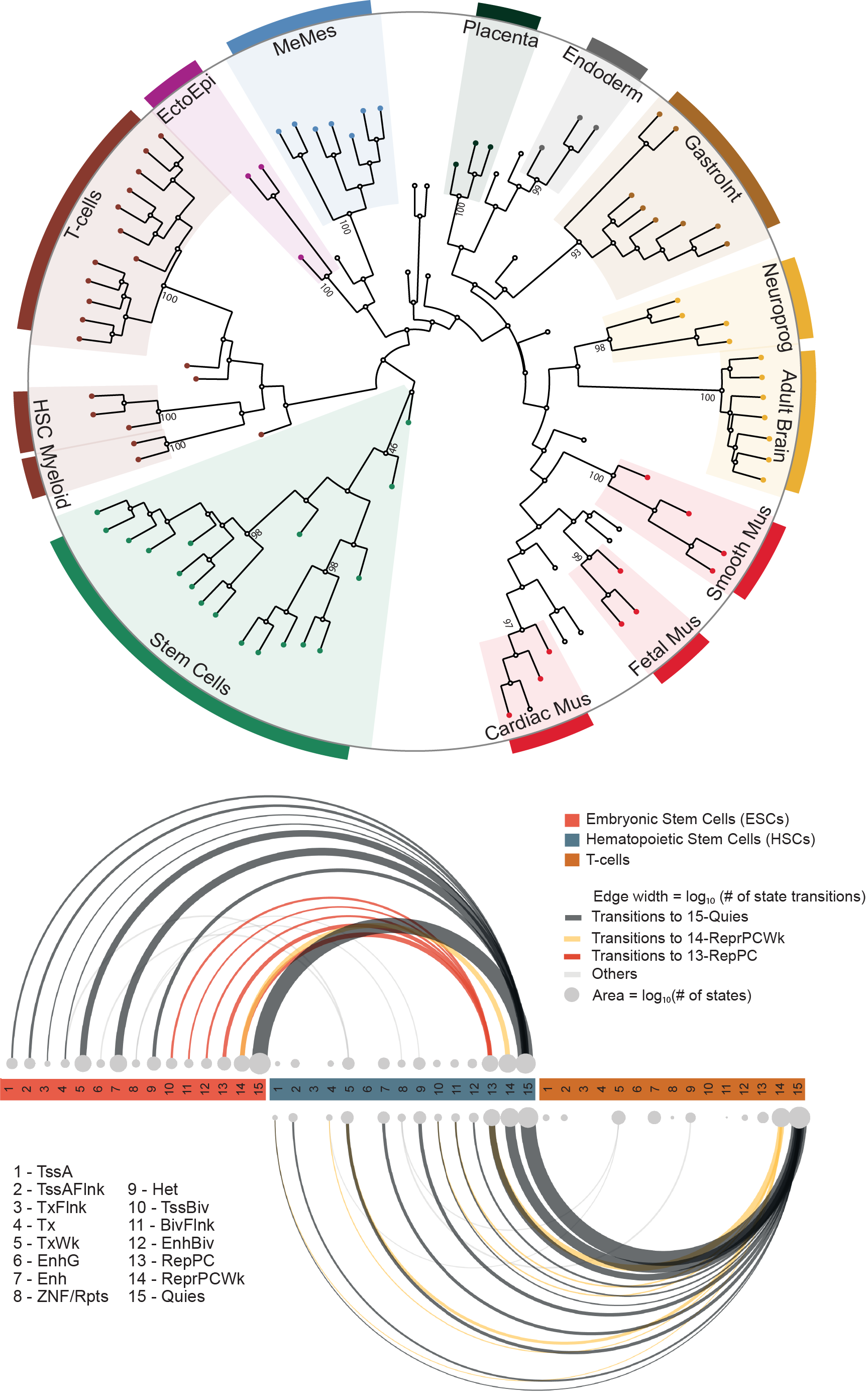
Tissue-specific expression of lincRNAs suggests developmental and cell-type-specific functions, yet tissue specificity was established for only a small fraction of lincRNAs. Here, by analysing 111 reference epigenomes from the NIH Roadmap Epigenomics project, we determine tissue-specific epigenetic regulation for 3,753 (69% examined) lincRNAs, with 54% active in one of the 14 cell/tissue clusters and an additional 15% in two or three clusters. A larger fraction of lincRNA TSSs is marked in a tissue-specific manner by H3K4me1 than by H3K4me3. The tissue-specific lincRNAs are strongly linked to tissue-specific pathways and undergo distinct chromatin state transitions during cellular differentiation. Polycomb-regulated lincRNAs reside in the bivalent state in embryonic stem cells and many of them undergo H3K27me3-mediated silencing at early stages of differentiation. The exquisitely tissue-specific epigenetic regulation of lincRNAs and the assignment of a majority of them to specific tissue types will inform future studies of this newly discovered class of genes.
Epigenomic footprints across 111 reference epigenomes reveal tissue-specific epigenetic regulation of lincRNAs.
Nature Communications, 6 (2015).
(article)
Created an online suite of tools to conduct epigenomic analyses. I tested, developed, and imporved analysis pipelines using internal or external developed softwares. Also, closely interacted with the developers to integrate analysis pipelines or data visualization methods in creating the Epigenome Toolset hosted in Genboree Workbench.
Using AngularJS and d3.js, I developed a data visualization tool that helps visualize the chromatin state transitions between samples. (Interative Tool with static dataset)
-
Bioinformatics Lead @ Baebies, Durham, NC, April 2018 - Present
-
Bioinformatics Postdoctral Fellow @ UTSW, Dallas, TX, June 2016 - April 2018
- Bioinformatics Research Assistant @ Baylor College of Medicine, Houston, TX, March 2012 - June 2016
-
Research Assistant @ UTSW, Dallas, TX, Oct 2008 - June 2011
- (2016-2017 Summer) One Day R Bootcamp @ UTSW, Dallas, TX
- (2012-2016 Spring) Computer-aided discovery methods @ Baylor College of Medicine, Houston, TX
- (2013 Fall) Epigenome Informatics Workshop @ ASHG, Boston, MA
- (2012-2013 Spring) Epigenome Informatics Workshop @ Houston, TX
- (Sep 2019) Machine Learning Introduction Nanodegree @ Udacity
- (Jun 2017) Bayesian Statistics @ Coursera - University of California, Santa Cruz
- (May 2016) Algorithmic Toolbox @ Coursera - University of California, Santa Cruz